Getting Started
Welcome to SNPio! This guide will help you get started with reading, filtering, encoding, analyzing, and visualizing genotype data for population genomic and phylogenetic analysis.
SNPio supports multiple standard formats — VCF, PHYLIP, STRUCTURE, and GENEPOP — and provides a unified GenotypeData object for downstream analysis using a consistent interface.
This guide covers:
Installation options (pip, conda, Docker)
File readers and formats
Core usage examples
Experimental modules like AMOVA and TreeParser
Installation
There are several ways to install SNPio:
Using pip (recommended with a virtual environment)
Using conda
Using Docker
(Advanced) Installing globally with pip (not recommended)
Installing with pip
python3 -m venv snpio-env
source snpio-env/bin/activate
pip install snpio
Tip
For reproducibility, consider saving your environment to a requirements file:
pip freeze > requirements.txt
Installing with conda
conda create -n snpio-env python=3.12
conda activate snpio-env
conda install -c btmartin721 snpio
Installing with Docker
docker pull btmartin721/snpio:latest
docker run -it btmartin721/snpio:latest
Tip
SNPio is designed for Unix-based systems (Linux, macOS). On Windows, use WSL (Windows Subsystem for Linux) for full compatibility.
Importing SNPio
Once installed, you can import SNPio modules as needed:
from snpio import (
VCFReader, PhylipReader, StructureReader, GenePopReader,
NRemover2, PopGenStatistics, GenotypeEncoder
)
Tip
Only import the readers or tools you plan to use. This helps avoid clutter, reduces memory usage, and avoids overwhelming your namespace with unused classes.
Reading Genotype Data
SNPio provides dedicated readers for each supported format. Each reader returns a GenotypeData object, which enables downstream filtering, encoding, and statistical analyses.
Here’s a minimal VCF loading example:
vcf = "snpio/example_data/vcf_files/phylogen_subset14K_sorted.vcf.gz"
popmap = "snpio/example_data/popmaps/phylogen_nomx.popmap"
gd = VCFReader(
filename=vcf,
popmapfile=popmap,
force_popmap=True,
prefix="snpio_example",
plot_format="png",
verbose=True
)
The GenotypeData Object
All SNPio readers return a GenotypeData object, which serves as a central container for your genomic data and metadata. Once you load your data, you can access several useful attributes directly from this object.
Key Public Attributes
Attribute |
Description |
|---|---|
|
A NumPy array |
|
A list of sample (individual) IDs. |
|
The total number of individuals. |
|
The total number of SNPs (loci). |
|
A |
|
A list of population IDs corresponding to each sample. |
|
A dictionary mapping each sample ID to its population ID. |
|
A dictionary mapping each population ID to a list of its member sample IDs. |
|
The total number of unique populations. |
|
A dictionary mapping each population ID to its sample count. |
|
A dictionary mapping each population ID to a list of its sample row indices. |
|
A boolean indicating if population data is present. |
|
A list of reference alleles for each locus (VCF-derived). |
|
A list of alternate alleles for each locus (VCF-derived). |
|
A list of concrete names for each locus (e.g., “chr1:1001”). |
|
A dictionary mapping sample IDs to their genotype sequences. |
|
A dictionary of the keyword arguments used to initialize the object. |
|
A boolean that is |
|
The root output directory path for generated files. |
|
The dedicated directory path for plots. |
|
The dedicated directory path for reports. |
|
A boolean NumPy array where |
|
A boolean NumPy array where |
|
A boolean NumPy array where |
|
The overall proportion of missing data in the alignment. |
|
A pandas Series with the missing data proportion for each locus. |
|
A pandas Series with the missing data proportion for each individual. |
|
A pandas Series with the heterozygosity rate for each locus. |
|
A pandas Series with the heterozygosity rate for each individual. |
|
A boolean NumPy array that is |
|
The approximate memory footprint of the |
|
A boolean array indicating which samples are retained after filtering. |
|
A boolean array indicating which loci are retained after filtering. |
You can use these attributes to inspect your data at any stage of your analysis pipeline. For example:
# Load data
gd = VCFReader(filename="my_data.vcf.gz", popmapfile="popmap.txt")
# Inspect the data
print(f"Loaded {gd.num_inds} individuals and {gd.num_snps} SNPs.")
print(f"Populations found: {list(gd.popmap_inverse.keys())}")
print(f"First 5 samples: {gd.samples[:5]}")
print(f"Genotype of first sample at first SNP: {gd.snp_data[0, 0]}")
Specifying Populations
You can restrict analysis to specific populations using include_pops and/or exclude_pops:
gd = VCFReader(
filename=vcf,
popmapfile=popmap,
prefix="snpio_example",
include_pops=["EA", "GU", "ON"],
exclude_pops=["MX", "YU"]
)
Tip
You can combine include_pops and exclude_pops to enforce fine-grained control. Just be sure they don’t overlap.
Note
The population IDs in include_pops and exclude_pops must not overlap. If they do, an error will be raised.
force_popmap Behavior
The force_popmap=True option attempts to align the popmap and alignment automatically by:
Removing unmatched samples
Sorting both to match order
Avoiding errors on slight mismatches
If set to False, exact correspondence is required between the popmap and alignment sample IDs.
Supported Missing Data Characters
All SNPio readers treat the following as missing genotypes:
“N”
“.”
“?”
“-”
Population Map File Format
Popmap files map sample IDs to population IDs. SNPio supports:
Whitespace- or comma-delimited files
Optional header row: SampleID, PopulationID
Example (no header):
Sample1,Population1
Sample2,Population1
Sample3,Population2
Example (with header):
SampleID,PopulationID
Sample1,Population1
Sample2,Population1
Sample3,Population2
Writing Genotype Data to File
All GenotypeData objects created by SNPio readers (e.g., VCFReader, PhylipReader, StructureReader, GenePopReader) support interoperable export methods. This allows you to convert between formats seamlessly, regardless of the original input type.
Available write methods:
write_vcf(filepath)- Export to bgzipped VCF formatwrite_phylip(filepath)- Export to sequential PHYLIP formatwrite_structure(filepath)- Export to STRUCTURE formatwrite_genepop(filepath)- Export to GENEPOP formatwrite_popmap(filepath)- Export population map file. Useful if samples were filtered or modified.
These methods are available directly from any GenotypeData object.
Examples
Export filtered data to a new VCF:
gd.write_vcf("output_data.vcf.gz")
Convert a VCF file to STRUCTURE format:
from snpio import VCFReader
gd = VCFReader(
filename="input.vcf.gz",
popmapfile="popmap.txt",
force_popmap=True,
prefix="converted_data"
)
gd.write_structure("converted_data.str")
Convert STRUCTURE → PHYLIP:
from snpio import StructureReader
gd = StructureReader(
filename="input_structure.str",
popmapfile="popmap.txt"
)
gd.write_phylip("converted.phy")
Convert GENEPOP → VCF:
from snpio import GenePopReader
gd = GenePopReader(
filename="example.gen",
popmapfile="popmap.txt"
)
gd.write_vcf("converted_from_genepop.vcf")
Notes
Output files can be compressed (e.g., .vcf.gz) or plain text.
The same GenotypeData object can be written to any supported format, regardless of input format.
SNPio handles internal data transformation automatically (e.g., allele formats, headers, sample ordering).
Tip
This interoperability is useful for converting legacy formats or preparing input for downstream tools that require a specific genotype format.
Next Steps
Now that you’ve successfully loaded data, you’re ready to filter, encode, and analyze genotype datasets using NRemover2, GenotypeEncoder, and PopGenStatistics.
Continue to the next section: Filtering with NRemover2.
Filtering Genotype Data with NRemover2
The NRemover2 class provides a flexible and efficient way to clean and preprocess your genotype dataset before downstream analysis. It supports filters for:
Missing data thresholds (per sample, locus, population)
Minor allele count (MAC) and frequency (MAF)
Singleton and monomorphic loci
Biallelic filtering
Locus thinning and linked loci pruning
Random subsetting
Filters can be chained and must be finalized with .resolve().
Basic Example
from snpio import NRemover2
nrm = NRemover2(genotype_data)
gd_filt = (
nrm.filter_missing_sample(0.75)
.filter_missing(0.75)
.filter_missing_pop(0.75)
.filter_mac(2)
.filter_monomorphic(exclude_heterozygous=True)
.filter_singletons(exclude_heterozygous=True)
.filter_biallelic(exclude_heterozygous=True)
.resolve()
)
# Save filtered VCF
gd_filt.write_vcf("filtered_output.vcf.gz")
Note
You must call .resolve() at the end of the filter chain to apply the filtering logic and return a new GenotypeData object.
Filtering with exclude_heterozygous
Many filters accept the exclude_heterozygous=True flag, which ensures that only homozygous genotypes are considered during filtering. For example:
filter_singletons(exclude_heterozygous=True) removes singletons where the minor allele is present in only one homozygous genotype. If the minor allele is present in a heterozygous genotype, it will not be counted as a singleton, but rather as a monomorphic site.
filter_biallelic(exclude_heterozygous=True) retains only sites with at least one homozygous reference and one homozygous alternate.
filter_monomorphic(exclude_heterozygous=True) removes sites with only one allele. A site with a heterozygous genotype and all homozygous reference genotypes will still be considered monomorphic if exclude_heterozygous=True is set, as it will not count the heterozygous genotype as a valid allele.
Tip
Use exclude_heterozygous=True when you want to apply conservative filters that rely only on clear homozygous calls.
Key Filtering Methods
Method |
Description |
|---|---|
|
Removes individuals with missing data above threshold. |
|
Removes loci with missing data above threshold. |
|
Removes loci where any population exceeds threshold missingness. |
|
Removes loci with MAC below threshold. |
|
Removes loci with MAF below threshold. |
|
Removes sites with only one allele. |
|
Removes loci where the minor allele is present in only one genotype. |
|
Retains loci with exactly two alleles. |
|
Retains one SNP within every size bp window. |
|
Keeps only one SNP per scaffold/chromosome within size bp. |
|
Selects a random subset of size loci. |
|
Finalizes and applies all filters. |
Note
thin_loci() and filter_linked() are only available for VCFReader input. They rely on chromosome and position metadata.
Threshold Grid Search
Use search_thresholds() to explore the effects of various filtering thresholds:
nrm = NRemover2(genotype_data)
nrm.search_thresholds(
thresholds=[0.25, 0.5, 0.75, 1.0],
maf_thresholds=[0.01, 0.05],
mac_thresholds=[2, 5],
filter_order=[
"filter_missing_sample",
"filter_missing",
"filter_missing_pop",
"filter_mac",
"filter_monomorphic",
"filter_singletons",
"filter_biallelic"
]
)
Tip
Grid search helps identify thresholds that retain maximum informative loci while reducing noise from missing or rare variants.
Note
search_thresholds() is incompatible with thin_loci(), filter_linked(), and random_subset_loci().
Filtering Visualizations
Missingness Reports
After filtering, you can inspect missing data levels:
gd_filt.missingness_reports(prefix="filtered")
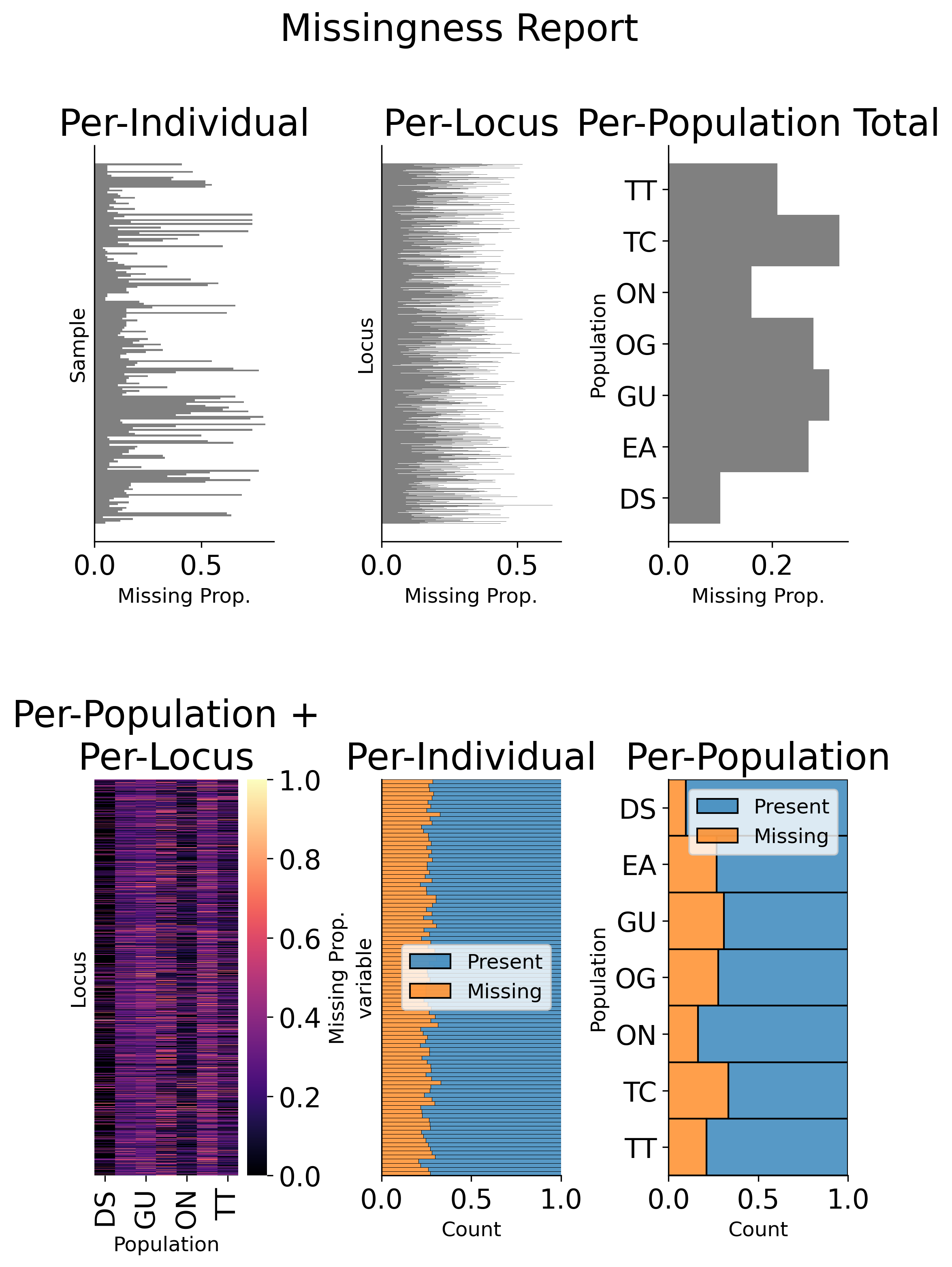
Figure: Per-sample, Per-locus, and Population-level missing data summaries.
Sankey Diagram
Visualize how many loci are retained vs. removed at each filter step:
nrm.plot_sankey_filtering_report()

Figure: Sankey diagram showing filter effects at each stage. Bands are proportional to the number of loci retained (green) or removed (red) at each step.
Note
The Sankey diagram only visualizes loci, not samples. It also must be run after .resolve().
Note
plot_sankey_filtering_report() does not render thin_loci(), filter_linked(), random_subset_loci(), or filter_missing_samples().
Best Practices
Always inspect missingness and allele distributions before analysis.
Use search_thresholds() to identify optimal filter thresholds for your dataset.
Call .resolve() before using filtered data downstream.
Combine visual tools (Sankey, missingness plots) to guide decisions.
Next Steps
Continue to Section 3: Encoding Genotype Data for Machine Learning and AI using the GenotypeEncoder class.
Encoding Genotype Data
SNPio provides the GenotypeEncoder class for converting genotype data into formats suitable for AI, machine learning, and numerical analyses. It supports:
One-hot encoding (multi-label binary)
Integer encoding (categorical)
0-1-2 encoding (for additive genetic models)
These formats can be used directly in machine learning pipelines or population genomic models.
Importing GenotypeEncoder
from snpio import VCFReader, GenotypeEncoder
gd = VCFReader(
filename="snpio/example_data/vcf_files/phylogen_subset14K_sorted.vcf.gz",
popmapfile="snpio/example_data/popmaps/phylogen_nomx.popmap",
force_popmap=True,
prefix="snpio_example",
verbose=True
)
encoder = GenotypeEncoder(gd)
Encoding Formats
One-Hot Encoding
gt_ohe = encoder.genotypes_onehot
Each base or IUPAC code is represented as a vector:
{
"A": [1.0, 0.0, 0.0, 0.0],
"C": [0.0, 1.0, 0.0, 0.0],
"G": [0.0, 0.0, 1.0, 0.0],
"T": [0.0, 0.0, 0.0, 1.0],
"N": [0.0, 0.0, 0.0, 0.0],
"W": [1.0, 0.0, 0.0, 1.0],
"R": [1.0, 0.0, 1.0, 0.0],
"M": [1.0, 1.0, 0.0, 0.0],
"K": [0.0, 0.0, 1.0, 1.0],
"Y": [0.0, 1.0, 0.0, 1.0],
"S": [0.0, 1.0, 1.0, 0.0],
}
Integer Encoding
gt_int = encoder.genotypes_int
Each genotype is mapped to a unique integer:
“A” = 0, “C” = 1, “G” = 2, “T” = 3
IUPAC heterozygotes (e.g., “W”, “R”) = 4-9
Missing values = -9
0-1-2 Encoding
gt_012 = encoder.genotypes_012
Encodes phased diploid data:
0 = Homozygous reference
1 = Heterozygous
2 = Homozygous alternate
-9 = Missing
Two-channel Allele Encoding
The two-channel alleles encoding provides separate matrices for each allele in diploid genotypes.
# Tuple of numpy arrays of shape (n_samples, n_variants)
gt_allele1, gt_allele2 = encoder.two_channel_alleles
Reversing the Encoding
All encodings are bi-directional. You can convert encoded data back to genotypes by setting the property:
encoder.genotypes_onehot = gt_ohe
encoder.genotypes_int = gt_int
encoder.genotypes_012 = gt_012
encoder.two_channel_alleles = (gt_allele1, gt_allele2)
This updates the original GenotypeData.snp_data matrix with the decoded genotype values.
Use Cases
AI and ML training data: Convert to numerical format for model input
Dimensionality reduction (e.g., PCA, t-SNE): Use 0-1-2 or one-hot formats
Visualization: Heatmaps, clustering, D-statistics, etc.
Tip
The encoder handles IUPAC ambiguity codes seamlessly and supports bidirectional conversion between genotypes and encoded formats.
Next Steps
Proceed to Section 4: Population Genetic Analyses with `PopGenStatistics` for computing Fst, heterozygosity, nucleotide diversity, D-statistics, PCA, and more.
Population Genetic Analysis
The PopGenStatistics class provides a unified interface to perform key population genetic analyses using your filtered GenotypeData object.
It supports:
Summary statistics: He, Ho, π, and pairwise Weir & Cockerham Fst (Weir and Cockerham, 1984)
Nei’s genetic distance (Nei, 1972) (observed, permutation p-values, bootstrap CIs)
D-statistics: Patterson’s (Green et al., 2010, Patterson et al., 2006), Partitioned (Eaton and Ree, 2013), and D-FOIL (Pease and Hahn, 2015)
Fst outlier detection (Beaumont and Nichols, 1996, Foll and Gaggiotti, 2008)
Principal Component Analysis (PCA)
(Experimental) AMOVA (Excoffier et al., 1992)
Basic Usage
from snpio import VCFReader, PopGenStatistics
gd = VCFReader(
filename="example.vcf.gz",
popmapfile="popmap.txt",
force_popmap=True,
prefix="analysis_output",
verbose=True
)
pgs = PopGenStatistics(gd)
# New API (returns a tuple)
summary_stats, allele_stats_df = pgs.summary_statistics(
fst_method="permutation", # "observed", "permutation", or "bootstrap"
n_reps=100,
n_jobs=-1,
save_plots=True
)
Calculates:
Observed heterozygosity (Ho)
Expected heterozygosity (He)
Nucleotide diversity (π)
Pairwise Fst (Weir & Cockerham, 1984)
Allele-frequency based summary statistics
Includes per-population tables and visualizations.
Allele Summary Statistics
SNPio provides a comprehensive allele-level diagnostic summary via the PopGenStatistics class. This module quantifies missingness, heterozygosity, allelic richness, minor allele frequencies (MAFs), and more — providing an overview of SNP dataset quality and diversity.
This is automatically called within the PopGenStatistics(…).summary_statistics() method.
Example
from snpio import VCFReader, PopGenStatistics
gd = VCFReader(
filename="example.vcf.gz",
popmapfile="popmap.txt",
force_popmap=True,
prefix="allele_stats"
)
pgs = PopGenStatistics(gd)
# New return order: (summary_stats, allele_stats_df)
summary_stats, allele_stats_df = pgs.summary_statistics(
fst_method="permutation",
n_reps=100,
n_jobs=-1,
save_plots=True
)
Output
Returns:
summary_stats: a dictionary of result DataFrames (e.g., Ho/He/π summaries, pairwise Fst matrices and, if applicable, p-values or bootstrap intervals).allele_stats_df: apd.DataFramewith >30 allele-level metrics.
Note
All proportions range from 0.0 to 1.0. The tuple return is ordered as (summary_stats, allele_stats_df).
Nei’s (1972) Genetic Distance
Computes pairwise Nei’s genetic distance between populations. Supports: observed distances, permutation-based p-values, and bootstrap confidence intervals.
nei_results = pgs.neis_genetic_distance(
method="permutation", # "observed", "permutation", or "bootstrap"
n_reps=1000,
n_jobs=-1
)
df_nei = nei_results["observed"]
df_pvals = nei_results.get("pvalues") # present if method == "permutation"
df_lower = nei_results.get("lower_ci") # present if method == "bootstrap"
df_upper = nei_results.get("upper_ci") # present if method == "bootstrap"
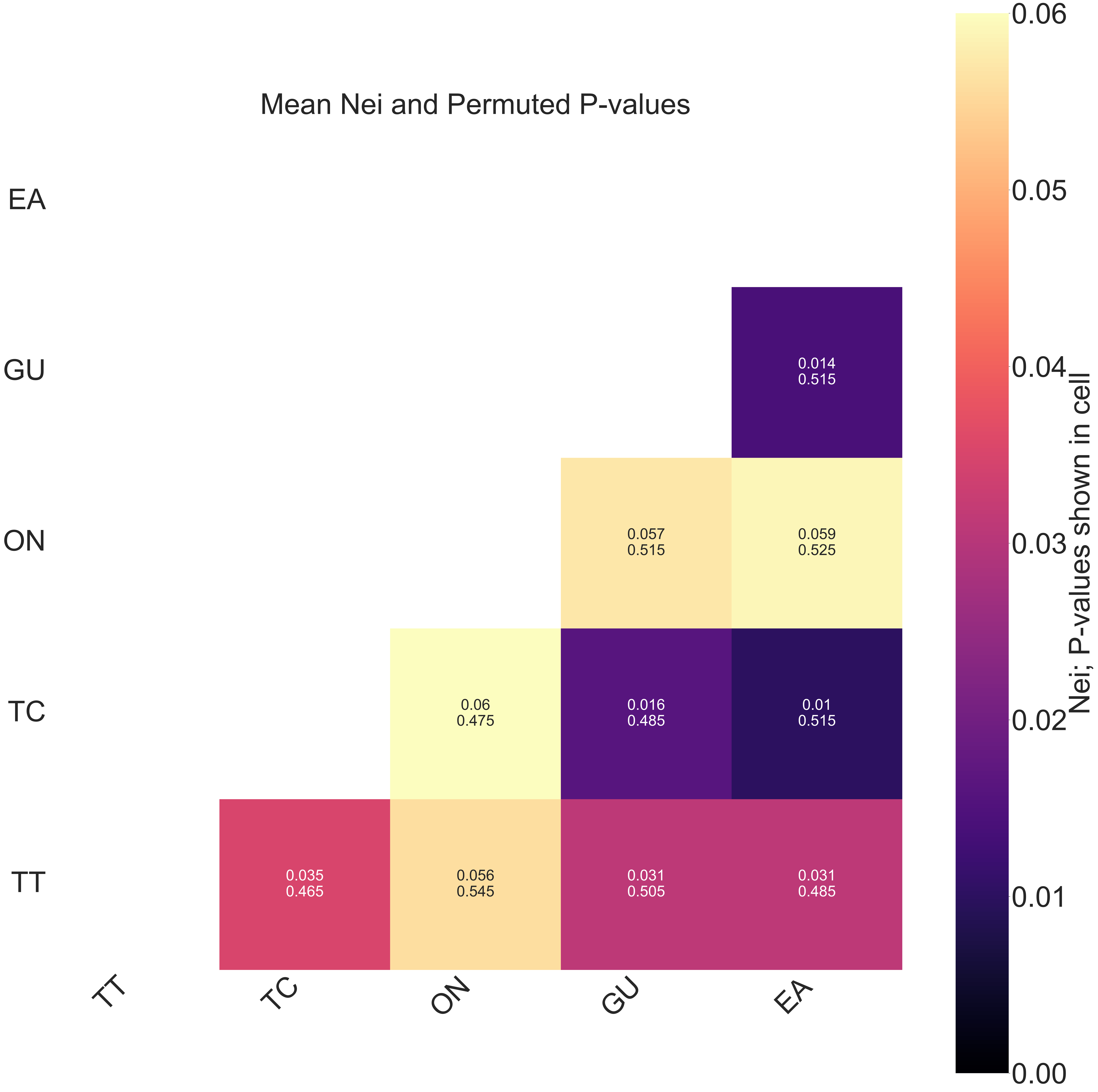
Figure: Heatmap showing Nei’s (1972) genetic distance between pairwise populations.
Tip
When plotted via SNPio’s built-ins and in the MultiQC report, diagonals are shown as 0.0 for distances and 1.0 for permutation p-values for readability.
Weir & Cockerham Fst (Distance Matrix)
Compute pairwise Weir & Cockerham Fst between populations. Supports: observed Fst, permutation-based p-values, and bootstrap confidence intervals.
fst_results = pgs.fst_distance(
method="permutation", # "observed", "permutation", or "bootstrap"
n_reps=1000,
n_jobs=-1,
palette="viridis",
suppress_plot=False
)
df_fst = fst_results["observed"]
df_fst_p = fst_results.get("pvalues") # if method == "permutation"
df_fst_l = fst_results.get("lower_ci") # if method == "bootstrap"
df_fst_u = fst_results.get("upper_ci") # if method == "bootstrap"
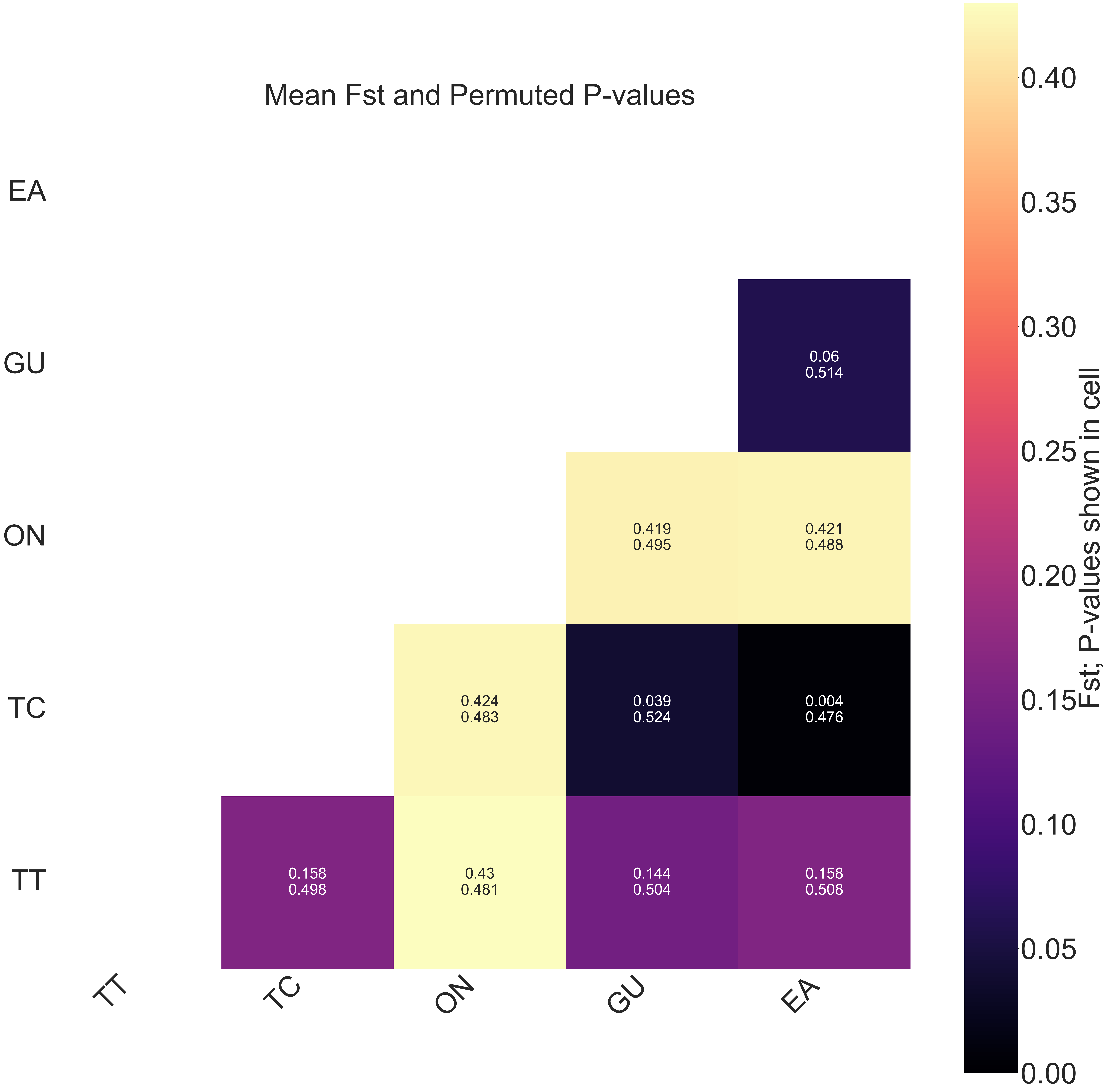
Figure: Pairwise Fst matrix. Lighter colors indicate higher differentiation.
D-Statistics (Patterson, Partitioned, DFOIL)
Calculate D-statistics for testing introgression among four (Patterson’s D), or five (Partitioned D, D-FOIL) populations. Supports bootstrap resampling, jackknife resampling, overall and per-combination statistics, and plotting.
Also supported are randomized or deterministic individual selection per population, and limiting the maximum number of individuals per population for balanced sampling and/or speed.
Basic Example
from snpio import VCFReader, PopGenStatistics
gd = VCFReader(
filename="example.vcf.gz",
popmapfile="popmap.txt",
prefix="dstat_analysis",
force_popmap=True,
)
pgs = PopGenStatistics(gd)
df, summary = pgs.calculate_d_statistics(
method="partitioned",
population1="P1",
population2="P2",
population3="P3a",
population4="P3b",
outgroup="Out",
num_bootstraps=1000,
max_individuals_per_pop=5,
individual_selection="random",
per_combination=True,
calc_overall=True,
save_plot=True,
use_jackknife=False,
block_size=500,
seed=42,
)
Fst Outlier Detection
Supports two methods:
Traditional permutations
DBSCAN clustering
# Permutation-based outliers (single DataFrame with unadjusted/adjusted
# p-values)
df_outliers = pgs.detect_fst_outliers(
n_permutations=1000,
correction_method="fdr_bh", # or "bonferroni", "holm", etc.
n_jobs=4,
seed=123
)
# DBSCAN-based outliers
df_dbscan = pgs.detect_fst_outliers(
use_dbscan=True,
correction_method="fdr_bh",
n_jobs=4,
min_samples=5
)
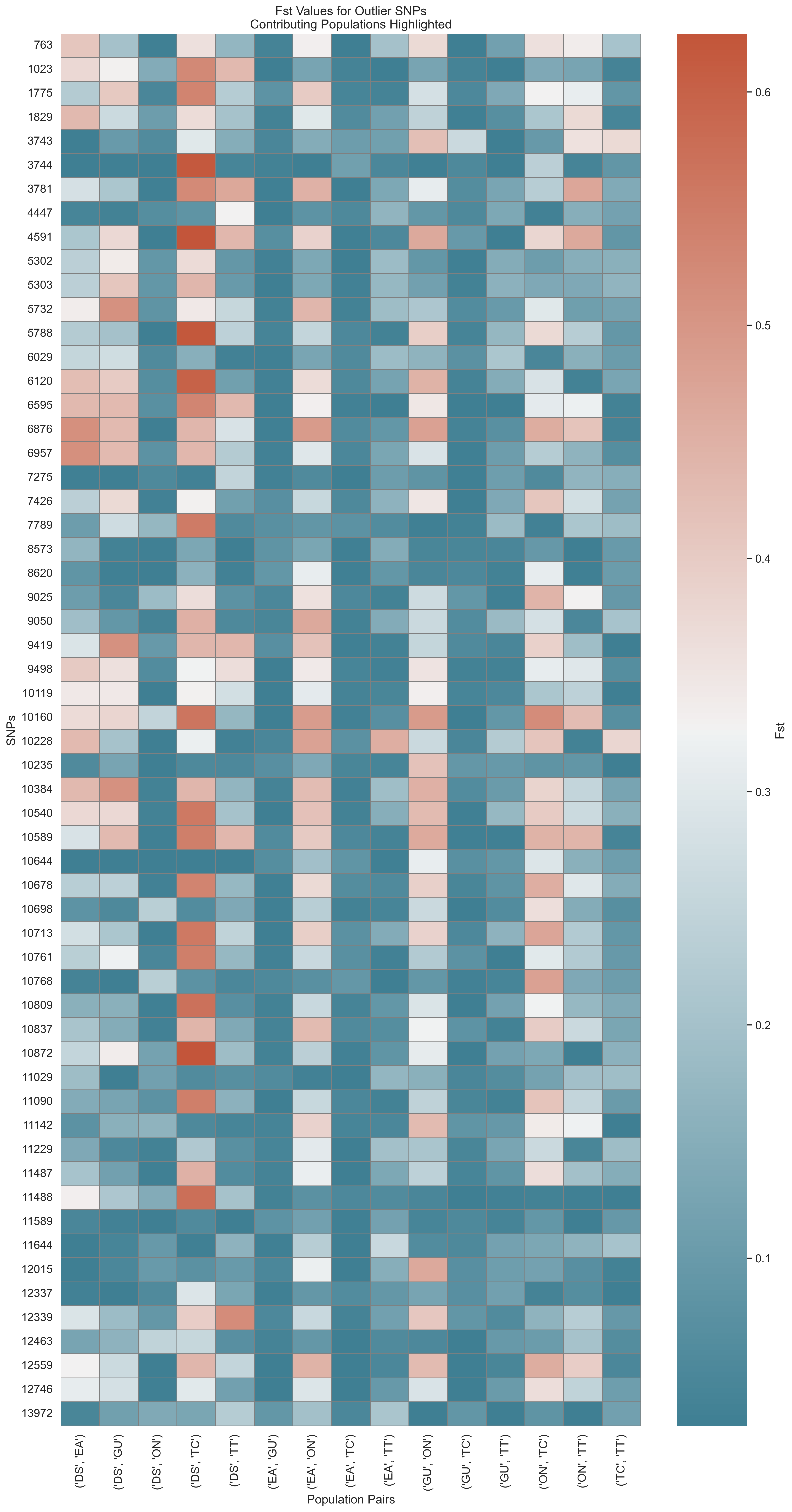
Figure: Outlier SNPs based on Fst between population pairs.
Note
detect_fst_outliers now returns one DataFrame. If a correction method is provided, adjusted p-values are included as columns.
AMOVA (Experimental)
amova_result = pgs.amova(
regionmap={
"EA": "East",
"GU": "East",
"TT": "East",
"DS": "West"
},
n_permutations=100,
n_jobs=1
)
Performs Analysis of Molecular Variance based on hierarchical population structure.
PopGenStatistics Method Summary
Method |
Description |
Algorithm(s) |
|---|---|---|
|
Calculates He, Ho, π, and pairwise Fst; returns |
Standard formulas; permutation/bootstrapping for Fst if requested |
|
Computes Nei’s pairwise distances. Returns dict with keys: |
Nei (1972) |
|
Computes Weir & Cockerham pairwise Fst. Returns dict like Nei’s: |
Weir & Cockerham (1984) |
|
Calculates Patterson / Partitioned / D-FOIL with bootstrap or jackknife support. |
ABBA–BABA framework with resampling |
|
Detects outlier loci via permutations or DBSCAN. Returns a single DataFrame (includes unadjusted/adjusted p-values when requested). |
Permutations or DBSCAN |
|
Runs PCA (KNN-imputes missing data, optional scaling, robust guards) and creates visualizations. |
scikit-learn PCA |
|
Performs AMOVA with optional SNP-wise permutations for p-values. |
Hierarchical variance partitioning |
Next Steps
Continue to Section 5: MultiQC Report Generation if you want to explore SNPio’s support for generating interactive MultiQC reports.
MultiQC Report Generation
SNPio includes built-in support for generating interactive MultiQC reports that summarize results from all major analysis modules. These reports are ideal for data exploration, publication-ready figures, and batch comparisons.
The SNPioMultiQCReport class automatically collects results from:
Filtering (NRemover2)
Summary statistics and Fst estimates (PopGenStatistics)
Genetic distance matrices (Nei’s, Weir & Cockerham’s)
D-statistics (Patterson, Partitioned, D-FOIL)
Fst Outlier detection (DBSCAN, permutations)
Genotype encodings (GenotypeEncoder)
PCA
Input/output summary tables (e.g., number of samples/loci retained)
Basic Example
After completing your filtering and statistical analyses:
from snpio import SNPioMultiQC
report = SNPioMultiQC.build(
prefix="my_report",
output_dir="results/multiqc",
overwrite=True,
)
Note
SNPioMultiQC should not be initialized directly. Instead, use it without parentheses followed by calling the build() method to generate the report from the current session’s results.
Tip
SNPioMultiQC should be called after all filtering and analysis steps are complete. It collects results from the current session and generates a comprehensive report of all filtering and analysis results.
This creates a self-contained .html report with interactive visualizations, significance summaries, and analysis metadata.
Tip
The MultiQC HTML report can be viewed in Firefox, Chrome, and possibly other web browsers. It is fully self-contained, meaning all data and visualizations are embedded in the HTML file.
Note
MultiQC support is available for all formats supported by SNPio (VCF, PHYLIP, STRUCTURE, GENEPOP).
Report Contents
The SNPio MultiQC report includes:
Filtering Summary (Sankey plot, missingness, sample/locus counts)
Summary Statistics (He, Ho, π)
Pairwise Fst Matrix (Weir & Cockerham)
Nei’s Distance Heatmap
D-statistics (histograms, significance bar plots)
Fst Outlier Heatmaps
PCA (colored by population)
Input Metadata Table (samples, loci, populations)
Custom Report Location
You can control the report path:
SNPioMultiQC.build(
prefix="Example Report",
output_dir="results/multiqc",
overwrite=True,
)
Tip
All generated reports are self-contained HTML files and require no internet connection to view.
Exporting Report Data
All visualizations in the report are backed by exportable CSV and JSON data, saved alongside the HTML file. This allows integration into downstream tools or publications.
Tip
The MultiQC HTML report is fully portable—just open the file in any browser or send it to collaborators.
Integration with Analysis Pipeline
You can integrate report generation as a final step in your SNPio pipeline:
# After all file I/O, filtering, encoding, and running PopGenStatistics
SNPioMultiQC.build(
prefix="Example Report",
output_dir="results/multiqc",
overwrite=True,
)
This ensures all results are captured in a single, interactive report for easy sharing and review.
Click Here to view an example MultiQC report generated by SNPio, showcasing the various analyses and visualizations available.
Next Steps
Continue to Section 6: Experimental Tree Parsing and Phylogenetic Analysis if you want to explore SNPio’s capabilities for working with phylogenetic trees and evolutionary models.
Tree Parsing and Phylogenetic Analysis (Experimental)
SNPio includes an experimental module, TreeParser, for working with phylogenetic trees in Newick or NEXUS formats. This class enables:
Reading and writing trees
Drawing and visualizing trees
Rerooting and pruning trees
Extracting subtrees by sample name
Getting distance matrices and rate models (Q-matrix, site rates)
Basic Example
from snpio import VCFReader, TreeParser
gd = VCFReader(
filename="snpio/example_data/vcf_files/phylogen_subset14K_sorted.vcf.gz",
popmapfile="snpio/example_data/popmaps/phylogen_nomx.popmap",
force_popmap=True,
prefix="snpio_example"
)
tp = TreeParser(
genotype_data=gd,
treefile="snpio/example_data/trees/test.tre",
siterates="snpio/example_data/trees/test14K.rates",
qmatrix="snpio/example_data/trees/test.iqtree",
verbose=True
)
Reading and Drawing Trees
tree = tp.read_tree()
tree.draw() # Visualize the tree
Saving Trees to File
Save in Newick format:
tp.write_tree(tree, save_path="output_tree.tre")
Save in NEXUS format:
tp.write_tree(tree, save_path="output_tree.nex", nexus=True)
Return tree as a string:
newick_string = tp.write_tree(tree, save_path=None)
Tree Editing and Analysis
Reroot tree using samples matching a pattern:
tp.reroot_tree("~EA") # Uses regex to match sample names containing 'EA'
Prune samples:
pruned = tp.prune_tree("~ON")
Extract subtree from a sample pattern:
subtree = tp.get_subtree("~EA")
Write pruned tree to file:
tp.write_tree(pruned, save_path="pruned_tree.tre")
Inspect tree statistics:
stats = tp.tree_stats()
print(stats)
Distance Matrix and Evolutionary Rates
Get full distance matrix between all nodes:
dist_matrix = tp.get_distance_matrix()
Access the rate matrix Q (from .iqtree file or Q-matrix):
print(tp.qmat)
Access site-specific substitution rates:
print(tp.site_rates)
Input File Notes
treefile: Required. Accepts Newick or NEXUS.
siterates: Optional. One-column file with rate per alignment site.
qmatrix: Optional. Can be parsed from .iqtree or provided as a CSV or TSV file with a 4×4 substitution rate matrix for A/C/G/T.
Experimental Status
Warning
TreeParser is still under active development. Interface and behavior may change in future versions.
Tip
Regular expressions can be used with ~ prefix in get_subtree() and prune_tree().
Example Use Case: Extracting a Subtree and Saving to Newick
subtree = tp.get_subtree("~EA")
newick = tp.write_tree(subtree, save_path=None)
print(newick)
Conclusion
TreeParser allows for interactive tree visualization and manipulation, fully integrated with SNPio’s genotype data. This feature is especially useful for combining genetic variation data with phylogenetic inference pipelines.
Thank you for using SNPio! We hope this guide helps you get started with reading, filtering, encoding, analyzing, and visualizing your genotype data effectively. For more advanced usage and examples, please refer to the documentation or explore the source code on GitHub.
For any questions or issues, feel free to open an issue on the SNPio GitHub repository.
References
Mark A. Beaumont and Richard A. Nichols. Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society B: Biological Sciences, 263(1377):1619–1626, 1996. doi:10.1098/rspb.1996.0237.
David A. R. Eaton and Richard H. Ree. Inferring phylogeny and introgression using radseq data: an example from flowering plants (\textit Pedicularis: orobanchaceae). Systematic Biology, 62(5):689–706, 2013. doi:10.1093/sysbio/syt032.
Laurent Excoffier, Peter E. Smouse, and Joseph M. Quattro. Analysis of molecular variance inferred from metric distances among dna haplotypes: application to human mitochondrial dna restriction data. Genetics, 131(2):479–491, 1992. doi:10.1093/genetics/131.2.479.
Matthieu Foll and Oscar Gaggiotti. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics, 180(2):977–993, 2008. doi:10.1534/genetics.108.092221.
Richard E. Green, Johannes Krause, and \textit et al. Briggs, Adrian W. A draft sequence of the neandertal genome. Science, 328(5979):710–722, 2010. doi:10.1126/science.1188021.
Masatoshi Nei. Genetic distance between populations. American Naturalist, 106(949):283–292, 1972. doi:10.1086/282771.
Nick Patterson, Alkes L. Price, and David Reich. Population structure and eigenanalysis. PLoS Genetics, 2(12):e190, 2006. doi:10.1371/journal.pgen.0020190.
James B. Pease and Matthew W. Hahn. Detection and polarization of introgression in a five-taxon phylogeny. Systematic Biology, 64(4):651–662, 2015. doi:10.1093/sysbio/syv023.
Bruce S. Weir and C. Clark Cockerham. Estimating f-statistics for the analysis of population structure. Evolution, 38(6):1358–1370, 1984. doi:10.2307/2408641.